
종목토론카테고리
게시판버튼
게시글 제목
'미국 FDA의 신약 심사 절차'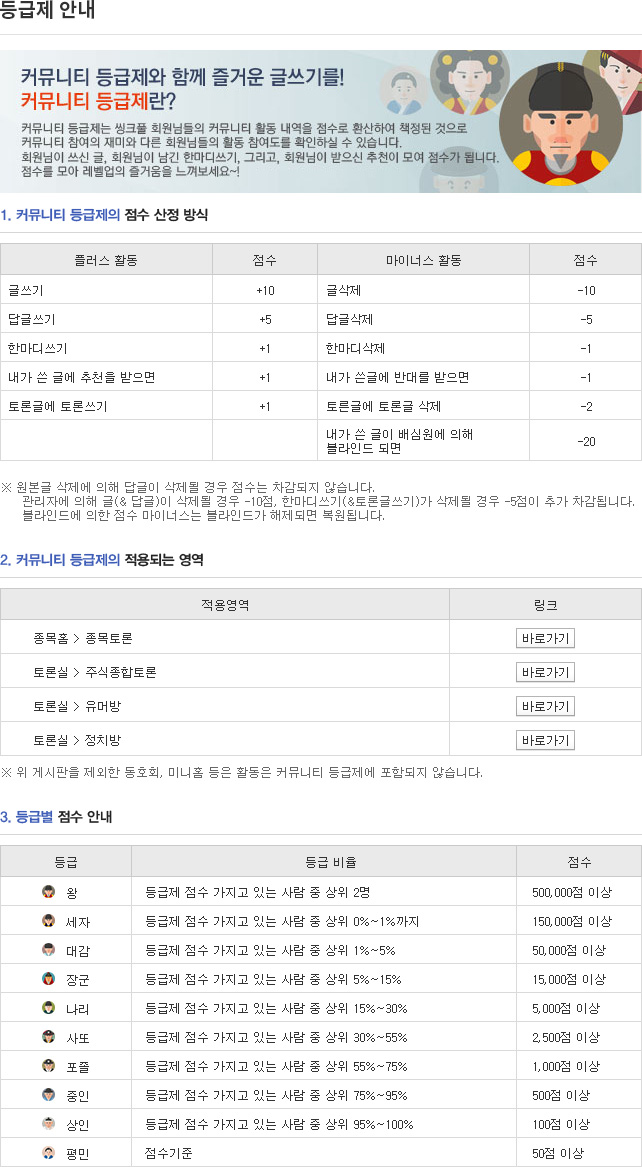
게시글 내용
미국 내 신약 심사, FDA 미팅 잘 활용해야"
안해영 박사, '미국 FDA의 신약 심사 절차' 강연
미국 신약 심사 과정에서 FDA와의 미팅은 각 단계별로 필요한 가이드라인 뿐만 아니라 규제 문제 등의 해법을 찾을 수 있는 기회가 될 것이라는 조언이다.
국내 최초 민간주도 바이오클러스터 우정바이오 신약클러스터의 출범을 기념하고자 우정바이오는 '제1회 우정바이오 신약클러스터 출범 기념 심포지엄'을 지난 1일 개최했다.
이날 심포지엄에 참가한 안바이오컨설팅 대표 안해영 박사는 '미국 FDA의 신약 심사 절차'라는 주제로 한국 회사들이 미국 시장에 진출하기 위해 필요한 정보를 공유했다.
| ▲ 안해영 박사가 심포지엄에서 발표를 진행하고 있다 |
미국 FDA에는 ▲Center for Tobcco Products(CTP) ▲Center for Veterinary Medicine(CVM) ▲Center for Biologics Evaluation and Research(CBER) ▲Center for Devices and Radiological Health(CDRH) ▲Center for Food safety and Applied Nutrition(CFSAN) ▲Center for Drug Evaluation and Research(CDER) 등 6개의 센터가 있다.
이 중 합성신약과 바이오신약의 심사는 CBER과 CDER이 맡고 있다.
안해영 박사에 따르면 FDA에서의 신약개발은 크게 ▲Pre-IND 단계 ▲IND 단계 ▲심사 단계, 총 3가지 단계로 나뉘게 된다.
Pre-IND 단계는 신약 후보의 탐구부터 비임상 시점까지가 포함되고, IND 단계는 임상 1상, 2상, 3상이 포함이 된다. 안 박사는 "사실 FDA는 Pre-IND 단계에서는 적극적으로 심사에 참여하지 않는다"고 말했다.
제약회사들이 동물 실험이 어느 정도 마무리되고 임상 시험을 준비하는 단계가 되었을 때 Pre-IND 미팅을 신청하면 FDA가 신약개발의 심사와 승인에 관여하기 시작한다는 설명이다.
이후 FDA는 각종 미팅을 통해서 회사들에게 가이드라인을 제공하고 Initial IND부터 임상에 필요한 모든 과정을 함께 하고 마지막으로 시장 진출을 위한 'Market Application Submission'을 심사하면서 신약 심사 과정에 참여한다.
안 박사는 "FDA와의 회의는 어떤 것도 강요적으로 진행되는 것은 없지만 미팅을 하면 많은 규제 문제들을 해결 받을 수 있다"며 "특히 FDA에는 세계에서 제출되는 모든 임상실험에 대한 자료를 가지고 있기 때문에 FDA의 심사 지식이나 경험을 받을 수 있는 미팅을 한국회사들 입장에서 안 할 이유가 전혀 없다"고 말했다.
이어 "미팅은 FDA에서 먼저 요청해 오지 않으니, 한국 회사들은 먼저 서면으로 미팅 요청해야 한다"고 설명했다.
| ▲ 안 박사가 신약개발에 있어 FDA와의 미팅의 중요성을 강조하고 있다 |
FDA와의 미팅은 ▲타입A ▲타입B ▲타입C 등 3가지 종류가 있다.
타입A는 주로 임상단계에서 문제가 발생했을 시 문제를 해결하고 다음 단계로 넘어갈 수 있는 내용을 나눈다.
타입B 회의에서는 주로 FDA의 조언을 구하는 회의로 Pre IND 미팅, Pre NDA/BLA 미팅, EOP1(End of Phase 1) 미팅, EOP2(End of Phase 2)/Pre-Phase 3 미팅이 타입 B 미팅에 속한다.
Pre IND 미팅은 정말 중요한 미팅이라고 안 박사는 설명했다. Pre IND 미팅의 목적은 임상과정 중 발생할 수 있는 문제점들을 FDA와 사전에 점검을 받는 것이고 FDA의 의견을 받을 수 있는 가장 명확한 장소라는 것.
안 박사가 공유한 FDA 조사결과에 따르면 Pre IND 미팅을 진행하고 희귀의약품 임상에 걸리는 기간은 7.1년으로 Pre IND 미팅 없이 진행한 희귀의약품 임상 소요시간 12.8년보다 무려 5.7년이라는 차이가 있었다.
타입C의 미팅은 A와 B에 속하지 않는 다른 모든 미팅들이 속한다.
안 박사는 "이 미팅들은 의무적인 것은 아니지만 강력하게 권고한다"고 말했다.
다음으로 초기 IND(Initial IND)의 경우, Initial IND를 FDA에 제출하면 FDA는 30일 동안 'Safety Review'를 진행한다. 이후 아무런 문제가 없은 FDA는 30일의 '진행허가편지(You May Proceed Letter)'를 보내고 문제가 발생하면 '임상보류편지(Clinical Hold Letter(CR))'를 보내게 된다.
CR은 크게 ▲Complete Clinical Hold ▲Partial Clinical Hold, 2가지로 나뉘게 된다.
Complete Clinical Hold는 신약후보물질이 미국에서 행하는 모든 임상 시험이 중지되는 것을 의미하고, Partial Clinical Hold는 다른 임상시험은 진행이 가능하나 특정 임상시험에서는 중지되는 것을 의미한다. 만약 임상을 진행 중인 회사가 30일 내로 FDA에 CR에 대한 답변을 보여야 한다.
신약신청서(NDA)나 의약품허가신청서(BLA)가 FDA에 제출이 된다면 처음 2개월의 접수기간(Filing Period)을 거쳐 10개월간의 심사기간(Review Period)을 가지게 된다. 총 12개월 단계를 걸쳐 최종 결정이 이뤄진다.
안 박사는 사전 NDA/BLA 미팅의 중요성 또한 언급했다. 사전미팅에서 회사는 가지고 있는 자료들을 FDA에 제공하고, 혹시라도 부족한 부분을 점검 받고 임상자료들을 어떤식으로 제출할 것인지 FDA와 의논하게 된다. 보통 FDA는 자료제출에 있기 2달 전부터 미팅을 가지라고 권고한다고 안 박사는 설명했다.
접수후 FDA 내에서는 신청서에 관한 심사가 이뤄진다. 결과는 ▲File the Application(접수완료) ▲Potentially Refuse to file the Application(잠재적 접수거부) ▲Refuse to file the Application(RTF)(접수거부) 등 3가지로 '잠재적 접수거부'의 의미는 부족한 자료를 일정기한 내 제출시 접수를 진행한다는 조건부 결과로, 회사가 시간 내에 제출하지 못하면 'RTF'로 이어지게 된다. 접수가 완료되면 회사는 60일 내로 확인받을 수 있고, 약물에 관한 심사가 시작하게 된다.
FDA에서 승인이 허가 된다는 연락이 오면 신청 회사들은 ▲Labeling ▲Post Marketing Requirement ▲Post Marketing Commitment 등의 협의가 최종 승인 전까지 이뤄져야 한다. 안 박사는 "PMR, PMC, Labeling 등에 관한 협의가 결정날까지 안되면 FDA 입장에서는 승인을 할 수 없기 때문에 회사들은 협의를 끝내는 것이 매우 중요하다"고 설명했다.
게시글 찬성/반대
- 2추천
- 1반대
내 아이디와 비밀번호가 유출되었다? 자세히보기 →





운영배심원의견
운영배심원의견이란
운영배심원 의견이란?
게시판 활동 내용에 따라 매월 새롭게 선정되는
운영배심원(10인 이하)이 의견을 행사할 수 있습니다.
운영배심원 4인이 글 내리기에 의견을 행사하게 되면
해당 글의 추천수와 반대수를 비교하여 반대수가
추천수를 넘어서는 경우에는 해당 글이 블라인드 처리
됩니다.
운영배심원(10인 이하)이 의견을 행사할 수 있습니다.
운영배심원 4인이 글 내리기에 의견을 행사하게 되면
해당 글의 추천수와 반대수를 비교하여 반대수가
추천수를 넘어서는 경우에는 해당 글이 블라인드 처리
됩니다.
댓글목록